

Translate this page into:
Eponymous Terminologies Related to Kaposi
*Corresponding author: Aravind Sivakumar, Department of Dermatology, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), Puducherry, India. aravinddermat@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Sivakumar A, Ramar SY. Eponymous Terminologies Related to Kaposi. Indian J Postgrad Dermatol. 2024;2:24-7. doi: 10.25259/IJPGD_101_2023
INTRODUCTION
Born in 1837 in Hungary of Jewish origin, Moritz Kaposi is a well-known dermatologist for his contributions to dermatology and oncology. Known initially as Moritz Kohn, he later changed his name to Kaposi after his birthplace, Kaposvar, in Hungary. Another explanation is based on his comments to colleagues that he changed his name to avoid confusion with five other similarly named physicians on the Vienna faculty. He studied at the University of Vienna and worked under the mentorship of Ferdinand Von Hebra, after which he succeeded as the professor and director of skin diseases. He made numerous contributions to the field of dermatology, including authoring books such as Lehrbuch der Hautkrankheiten (Textbook of Skin Disease) along with Hebra. He later authored other books, such as Pathology and Therapy of Skin Disease and Atlas of Skin Diseases.[1] He is well remembered for the descriptions of numerous skin conditions that bear his name, described below.
NAMED CONDITIONS AND SIGNS AFTER KAPOSI
Kaposi sarcoma (KS)
Initially termed ‘idiopathic pigmented sarcoma of the skin’ and it was first described in 1872 by Kaposi in five patients with this condition. It is caused by human herpes virus-8 (HHV8) also termed KS herpes virus. It is classified into various clinical types such as classical type, endemic or African type, epidemic or HIV-associated KS and KS associated with immunosuppression or solid organ transplantation. Mostly seen in older adults, it also affects young children, especially in the case of endemic KS. It can be polymorphic in presentation, presenting as asymptomatic or tender bluish-red macules [Figure 1a], plaques or nodules usually affecting the extremities and involving the mucosa. Dermoscopic features of KS include white dots and lines, white clods four-dot clods, collarette sign [Figure 1b], serpentine vessels and polychromatic lustre termed as rainbow pattern.[2,3] Histologically KS sarcoma is characterised by a proliferation of irregular, jagged vascular channels, which partly surround pre-existing blood vessels in some areas. This characteristic appearance of a small vessel protruding into an abnormal vascular space has been termed ‘promontory sign’. Treatment depends on the type of KS and includes surgical, chemotherapy and radiotherapy. While small, localised lesions can be excised, larger multifocal lesions respond well to cytotoxic agents such as vinblastine, paclitaxel and doxorubicin with radiotherapy if required. HIV/AIDS-related KS responds well to highly active antiretroviral therapy.[4]

- (a) Violaceous macules of Kaposi sarcoma in a patient with AIDS. (b) Reddish pink structureless areas with four dot clods and collarette sign in Kaposi sarcoma (Dermlite DL4, ×10).
Kaposi varicelliform eruption
Kaposi varicelliform eruption also termed eczema herpeticum, is an acute viral eruption most caused by herpes simplex virus in individuals with pre-existing dermatoses. It can also be caused by the coxsackie virus (eczema coxsackium), varicella-zoster virus, vaccinia virus (eczema vaccinatum) and other viruses. The underlying conditions are mainly atopic dermatitis, pemphigus foliaceous, Darier’s disease, benign familial pemphigus, allergic contact dermatitis and Sezary syndrome. It presents as multiple vesiculopustular lesions with central umbilication, forming punched-out polycyclic erosions [Figure 2] with haemorrhagic crusting. It is associated with fever and systemic symptoms based on visceral involvement, such as hepatic, neurological, pulmonary and renal disease. Diagnosis can be confirmed by cytological methods with Tzanck smear demonstrating multinucleated giant cells and by more sensitive methods such as viral polymerase chain reaction and viral culture. Treatment includes antivirals, mainly acyclovir dosed intravenously, and optimisation of the underlying condition.[5,6]
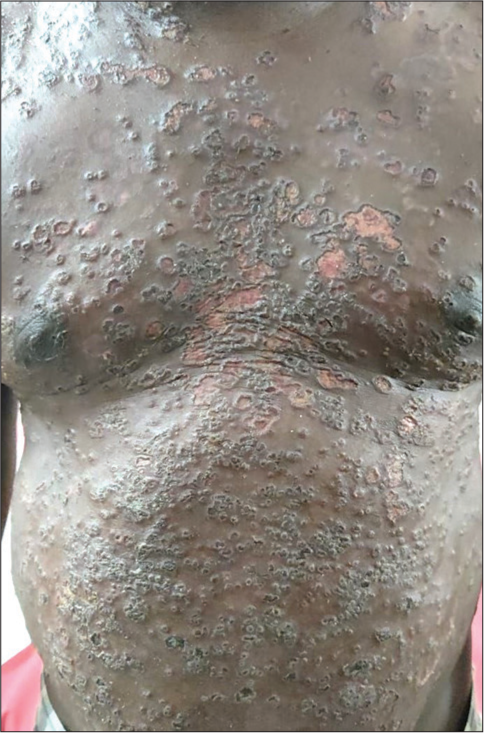
- Punched out polycyclic erosions of eczema herpeticum in pemphigus vulgaris.
Kaposi-Irgang disease
In 1883, Kaposi found the association of cutaneous nodules with systemic lupus erythematosus (SLE). Later, Arnold termed this particular entity as lupus profundus or Kaposi-Irgang disease. Lupus erythematosus profundus predominantly affects the deeper layers of skin including subcutis, and it is termed lupus panniculitis if superficial skin changes are absent. It presents as tender, erythematous indurated nodules or plaques mainly over the face, proximal extremities and back after the onset of SLE or sometimes can precede the development of SLE. It is often refractory to conventional agents such as hydroxychloroquine and other immunosuppressant’s and often requires aggressive therapy to prevent lipoatrophy, calcinosis and disfigurement.[7]
Pseudo Kaposi sarcoma
Furthermore referred to as acroangiodermatitis of mali or acral capillary angiomatosis, it is a form of reactive vascular proliferation secondary to venous stasis. It closely mimics KS hence the name and often occurs in the setting of chronic venous insufficiency, arteriovenous anomalies and paralysed or amputated limbs. The variants of acroangiodermatitis include Stewart–Bluefarb syndrome occurring in cases of congenital venous malformation, pregnancy-associated gravitational purpura and cases of haemodialysis associated with chronic renal disease. It presents as bluish macules, nodules and feet of the distal extremities mainly the feet. Clinically and histologically mimics KS and can be distinguished by the absence of nuclear atypia, CD34-positive spindle cell proliferation and negative immunostaining for HHV-8. Treatment is directed at correcting the underlying vascular abnormality by conservative methods such as compression therapy or surgical procedures.[8]
Kaposiform haemangioendothelioma
It was first described by Zuckerberg et al. in 1993 as a rapidly aggressive tumour with a locally infiltrative growth mimicking KS, hence named Kaposiform haemangioendothelioma.[9] It usually occurs in the 1st year of life and needs to be differentiated from other vascular tumours of infancy, such as infantile haemangioma, tufted angioma and vascular malformations. It is usually sporadic with few cases demonstrating somatic mutations in GNA/GNAQ. Clinically, it presents as indurated purplish firm papules, nodules or plaques. High index of suspicion is needed to look for red flag signs such as a sudden increase in size/swelling, increased tenderness, warmth, erythema or purpura, suggesting underlying Kasabach-Merritt syndrome (KMS). Other local complications due to aggressive growth include pain and functional limitations of limb movements, lymphoedema and compression of vital structures. Histology reveals the presence of confluent and well-defined nodular aggregates of endothelial spindle cells forming slit-like vascular lumina and lymphovascular channels comprising red blood cells with extravasation. Immunostaining is positive for endothelial markers (CD31 and CD34) and lymphovascular markers (VEGFR-3 and D2-40) but negative for Glut-1 and HHV-8, differentiating from infantile haemangioma and KS, respectively. Treatment is mainly aimed at preventing and treating KMS and includes a combination therapy of any of the following agents: corticosteroids, sirolimus, vincristine and propranolol.[10]
Kaposiform lymphangiomatosis
Kaposiform lymphangiomatosis is a newly described lymphatic disorder in 2014, comprising kaposiform spindled cells. It needs to be differentiated from kaposiform haemangioendothelioma. Kaposiform lymphangiomatosis occurs in older children, usually intrathoracic in location, with poor response to therapy, causing increased morbidity and mortality. Histologically, it comprises clusters of spindled cells arranged in parallel poorly defined strands or sheets. Complications include bleeding manifestations such as Kasabach-Merritt phenomenon but less severe and pericardial or pleural effusion in case of intrathoracic disease.[11]
Kaposi-Stemmer sign
Patients with chronic lymphoedema developed progressive swelling of the extremities, which is non-pitting in early stages, progressing to thickening and marked hyperkeratosis with papillomatosis referred to as elephantiasis nostras verrucosa cutis. Clinical signs of lymphoedema can help to distinguish it from other causes of leg oedema. KaposiStemmer sign refers to the inability to pinch the base of the second toe [Figure 3] and occurs due to the progressive thickening and fibrosed skin beneath. It is a usual clinical marker that is specific for diagnosing lymphoedema and can be confirmed further by lymphoscintigraphy.[12]
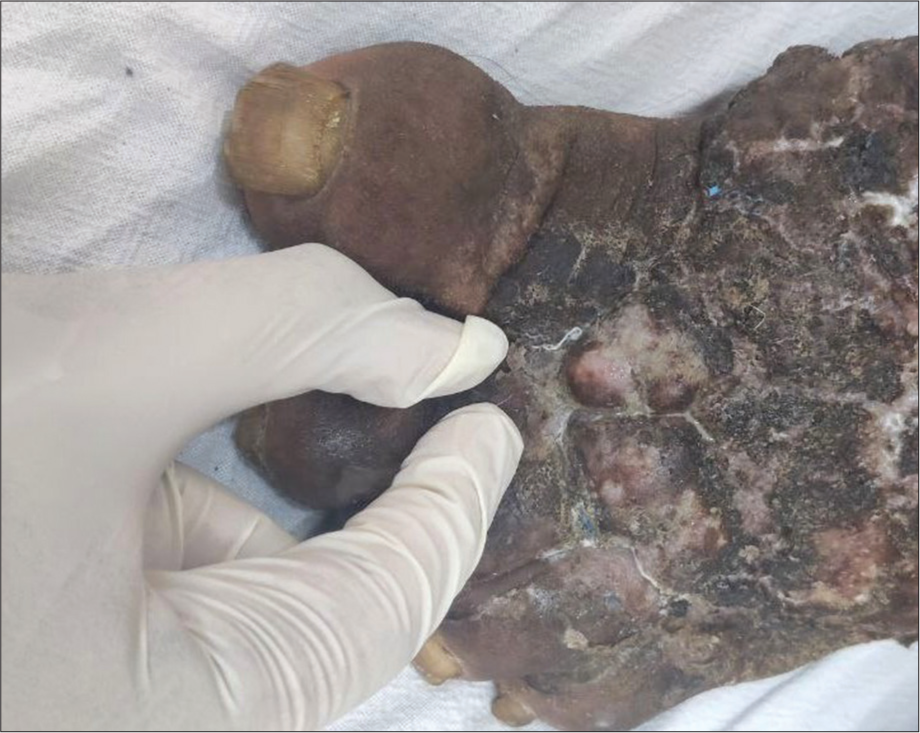
- Kaposi-stemmer sign in filarial lymphoedema.
Kaposi dermatitis
Xeroderma pigmentosum was initially described by Hebra and Kaposi in 1874, hence also referred to as Kaposi dermatitis and later renamed to Xeroderma pigmentosum, meaning dry pigmented skin. It belongs to the group of DNA repair disorders characterised by prominent photosensitivity manifesting as increased sunburn and progressive freckling with an increased predisposition to develop precancerous lesions and skin cancers. Associated systemic involvement includes progressive neurodegeneration, eye changes and hearing loss. Diagnosis is mainly clinical and confirmed by measurement of unscheduled DNA synthesis in skin fibroblasts. Management mainly consists of photoprotective measures for the prevention and treatment of skin cancers.[13]
CONCLUSION
Kaposi died at the age of 65 years in the year 1902 at Vienna. He is still remembered for his rich legacy in the field of dermatology for the conditions that bear his name as enumerated above. Hence, the dermatologist must be aware of these conditions as a tribute to Kaposi.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Moritz Kaposi: A Notable Name in Dermatology. JAMA Dermatol. 2015;151:867.
- [CrossRef] [PubMed] [Google Scholar]
- Dermoscopic Findings of Kaposi Sarcoma and Dermatopathological Correlations. Australas J Dermatol. 2020;61:e46-53.
- [CrossRef] [PubMed] [Google Scholar]
- The Dermoscopic Rainbow Pattern-A Review of the Literature. Acta Dermatovenerol Croat. 2019;27:111-5.
- [Google Scholar]
- Genotypic and Clinicopathological Characterization of Kaposi's Sarcoma-associated Herpesvirus Infection in Japan. J Med Virol. 2010;82:400-6.
- [CrossRef] [PubMed] [Google Scholar]
- Kaposi's Varicelliform Eruption. Indian J Dermatol Venereol Leprol. 2007;73:65.
- [CrossRef] [PubMed] [Google Scholar]
- Kaposi Varicelliform Eruption in Patients with Darier Disease: A 20-Year Retrospective Study. J Am Acad Dermatol. 2015;72:481-4.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical Characteristics of Lupus Erythematosus Panniculitis/Profundus: A Retrospective Review of 61 Patients. JAMA Dermatol. 2020;156:1264-6.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic Workup of Acroangiodermatitis of Mali (pseudo-Kaposi sarcoma) Demasking Metastasized Epithelioid Angiosarcoma. J Dtsch Dermatol Ges. 2020;18:1475-7.
- [CrossRef] [Google Scholar]
- Kaposiform Hemangioendothelioma of Infancy and Childhood. An Aggressive Neoplasm Associated with Kasabach-Merritt Syndrome and Lymphangiomatosis. Am J Surg Pathol. 1993;17:321-8.
- [CrossRef] [PubMed] [Google Scholar]
- Kaposiform Hemangioendothelioma: Current Knowledge and Future Perspectives. Orphanet J Rare Dis. 2020;15:39.
- [CrossRef] [PubMed] [Google Scholar]
- Kaposiform Lymphangiomatosis: A Distinct Aggressive Lymphatic Anomaly. J Pediatr. 2014;164:383-8.
- [CrossRef] [PubMed] [Google Scholar]
- The Diagnostic Unreliability of Classic Physical Signs of Lymphedema. J Vasc Surg Venous Lymphat Disord. 2019;7:890-7.
- [CrossRef] [PubMed] [Google Scholar]
- Xeroderma pigmentosum (Kaposi): Report of a Case, with Special Reference to Clinical Features and Pathogenesis. Br J Dermatol. 1926;38:241-52.
- [CrossRef] [Google Scholar]




