

Translate this page into:
Localised Epidermolysis Bullosa Simplex in Mono-Zygotic Twins

*Corresponding author: Rahul Suresh Nayak, Department of Dermatology, Hassan Institute of Medical Sciences, Hassan, Karnataka, India. drrahulnayak19@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Nayak RS, Nirvanappa VK. Localised Epidermolysis Bullosa Simplex in Mono-Zygotic Twins. Indian J Postgrad Dermatol. doi: 10.25259/IJPGD_232_2024
Abstract
Epidermolysis bullosa simplex is a rare blistering disorder without any curable treatments, mainly inherited in an Autosomal Dominant fashion. It is even rarer in twins. It is characterised by cutaneous fragility and blister formation after minimal trauma. It typically presents with localised, mild, acral blistering and normal life expectancy. Its localised presentation, confined to the palms and soles and association with punctuate callosities, may lead to misdiagnosis of Palmoplantar warts. We report two cases of localised EBS occurring in mono-zygotic twins with no family history.
Keywords
Epidermolysis bullosa
Epidermolysis bullosa simplex
Mechanobullous disorders
Punctate callosities
Twins

INTRODUCTION
Epidermolysis bullosa (EB) is a rare genodermatosis, characterised by mucocutaneous fragility and blister formation after minor friction or trauma.[1,2] EB is classified based on the level of split and is divided into four types, that is EB Simplex (EBS), Junctional EB (JEB), Dystrophic EB (DEB) and mixed EB (Kindler syndrome).[3] EBS is the most common type of EB, accounting for 75–85% of cases of EB in the Western world.[4] EBS is usually caused by pathogenic variants in the keratin genes (KRT5 and KRT14) with resultant formation of a cleavage plane at the level of the basal keratinocytes. Localised EBS (formerly known as Weber–Cockayne EBS), usually associated with little or no extracutaneous involvement, is the mildest and most common form of EBS.[5] In this report, we describe two cases of Localised EBS occurring in mono-zygotic twins with no family history.
CASE REPORT
Two 16-year-old monozygotic twin females presented with recurrent painful blisters on their hands and feet, starting at 3–4 years of age. The blisters initially contained clear fluid, later becoming cloudy or red-black before rupturing and forming black scabs that eventually healed, leaving white patches. Over time, thickened, raised lesions developed on the soles. The blisters worsened in hot weather, and both patients experienced excessive sweating in the palms and soles. No family history of similar conditions was noted, though the parents were consanguineous.
On examination, both patients had tense bullae with cloudy fluid, brownish crusts on their feet and soles and hypopigmented macules on the palms and hands. Hyperkeratotic calluses with brown crusts were also seen on the soles. Nail changes included onychodystrophy and subungual hyperkeratosis. General examination was normal, with no mucosal or hair involvement [Figures 1 and 2].
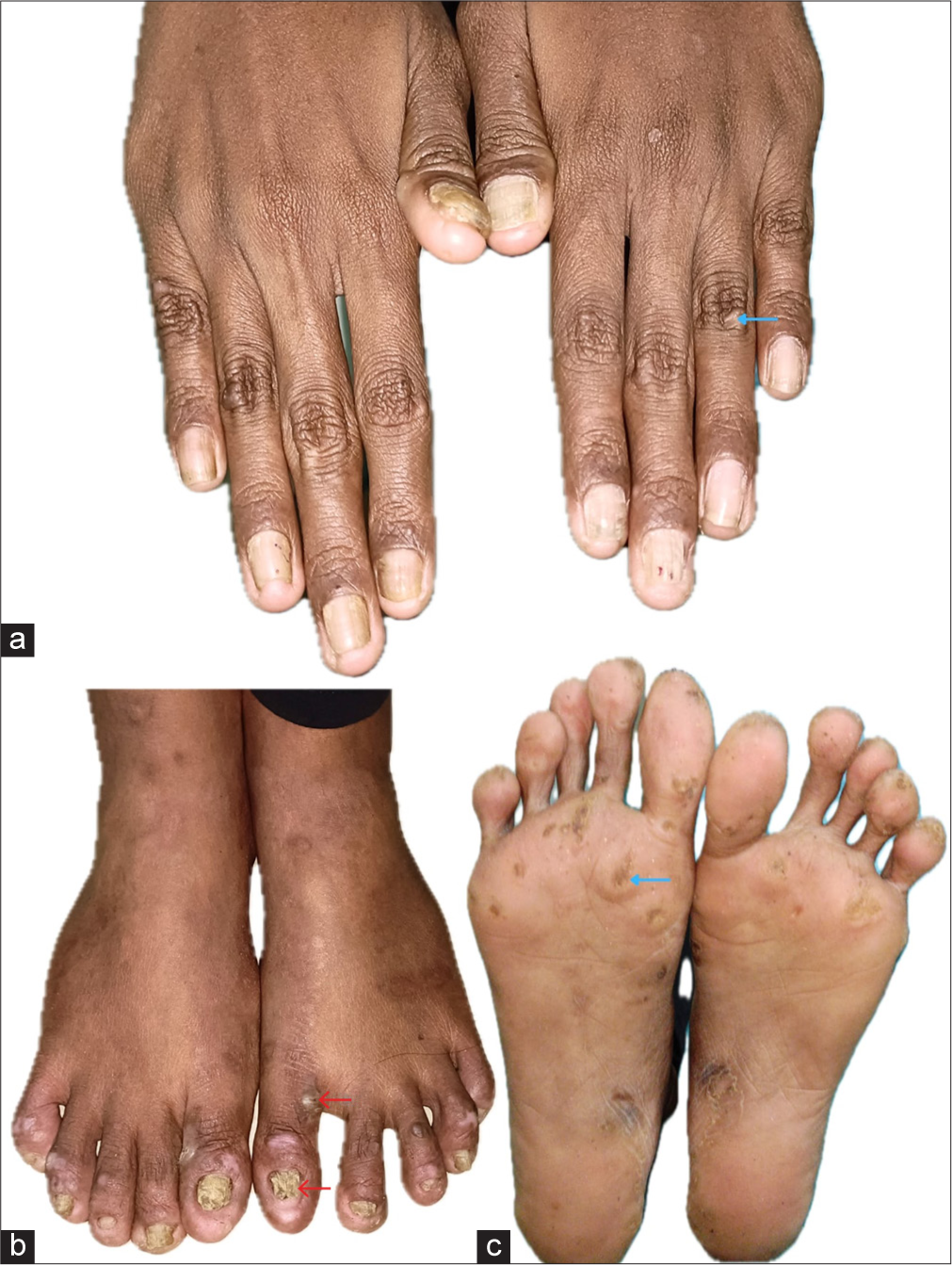
- Clinical presentation of Patient 1. (a) Hypopigmented macules on the hands (blue arrow). (b) Tense bullae with cloudy fluid and overlying brownish crusts on the feet (blue arrow). Nail changes, including onychodystrophy and subungual hyperkeratosis (red arrow). (c) Hyperkeratotic calluses with brown crusts on the soles (blue arrow).
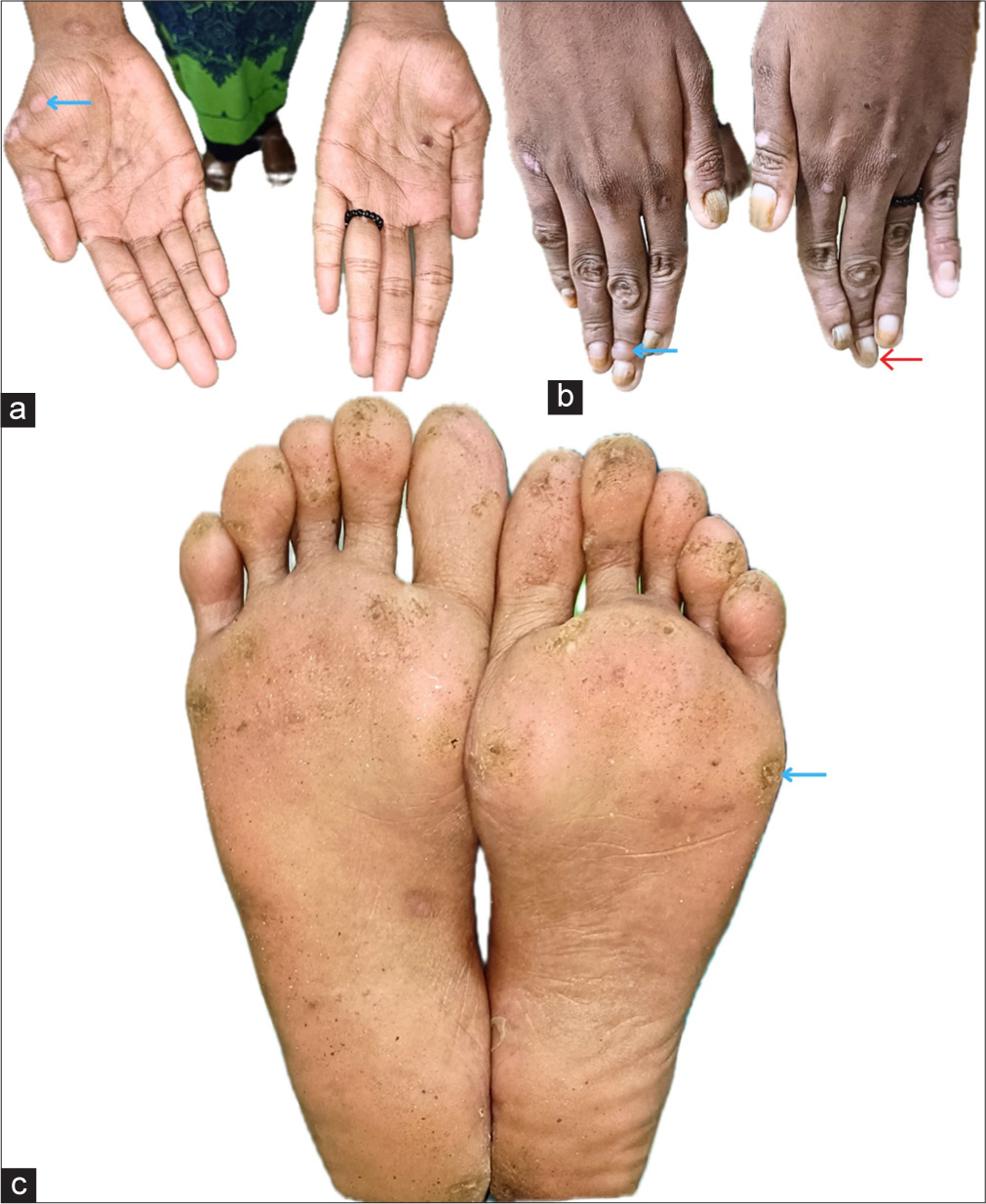
- Clinical presentation of Patient 2. (a) Hypopigmented macules on the palms (blue arrow). (b) Tense bullae with clear fluid over the finger dorsa (blue arrow). Nail changes, including onychodystrophy (red arrow). (c) Hyperkeratotic calluses with brown crusts on the soles (blue arrow).
Laboratory tests, including complete blood count and liver/renal function, were normal. Histopathology revealed a suprabasal cleft in the epidermis without inflammation [Figure 3]. Direct immunofluorescence was negative. Due to high costs, electron microscopy and genetic testing were not performed.
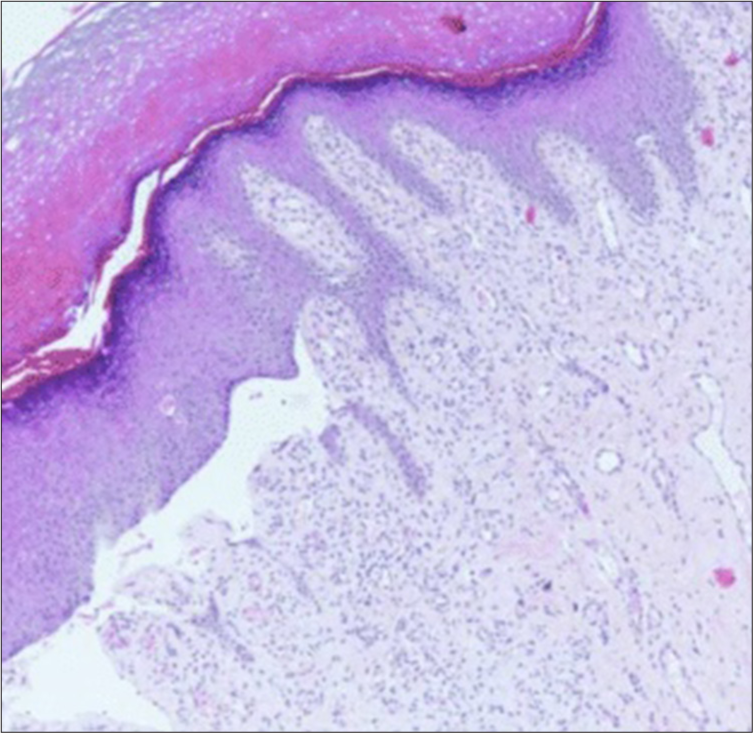
- Histopathology: Suprabasal cleft in the epidermis, without inflammation (Haematoxylin and eosin stain, 10x).
The patients were counselled about the chronic nature of the condition and lack of curative treatments. Topical fusidic acid cream was prescribed for lesions, and protective footwear was recommended. Parental consent for anonymised photographs was obtained for publication.
DISCUSSION
It is acknowledged that hereditary elements and genetic mutations can contribute to the predisposition to EB group of disorders. The defining characteristics of EB encompass mucocutaneous blisters resulting from tissue injury which leads to erosion and ulceration, typically resulting from frictional trauma. The EB group of disorders is classified based on the level of split and is divided into four types – EBS, JEB, DEB and mixed EB (Kindler syndrome). According to epidemiological data, EBS emerges as the predominant form of EB, constituting over 70% of all EB cases. According to the most recent classification, there are 14 EBS subtypes. The three primary subtypes include Localised EBS (Weber–Cockayne), intermediate (generalised intermediate/Koebner) and severe (Dowling-Meara).[2,3] Common features frequently observed in EBS include symptoms that manifest early in life (during early childhood), and blistering symptoms are often provoked by elevated air temperatures. In our case report, these symptoms were aggravated by hyperhidrosis. In EBS, occurrences of mucosal symptoms and nail damage are infrequent. It is inherited in an autosomal dominant manner in most cases. In some cases, it can be inherited in an autosomal recessive manner. Electron microscopy is the gold standard diagnostic method for EB. It provides a more precise assessment of blistering levels compared to biopsy and immunofluorescence examinations. In addition, electron microscopy offers the capability to visualise cells and subcellular structures that conventional (ultrastructural) microscopes cannot access. In this instance, direct immunofluorescence is utilised to eliminate potential differential diagnoses of EB. The findings from the direct immunofluorescence analysis showed that no presence of immunoglobulin G and C3 deposits in the basement membrane zone, thus excluding the diagnosis of bullous pemphigoid and EB acquisita. Another supplementary examination for determining the type of EB is genetic testing. Research indicates various genetic mutation causes for each EB type. Most common mutation in EBS is the KRT5 gene, which codes for Keratin, type II cytoskeletal 5 protein.[2] A novel de novo c.2T>C (p.M1T) variant in KLHL24 was identified by Xu et al. (2021) and linked to EBS in twin boys, emphasising the importance of early genetic diagnosis for effective management.[6] Most EB therapy is carried out in a supportive manner. Treatments, including wound care, infection control, adequate nutrition and genetic counselling, are available, alongside the potential for novel therapeutic approaches like gene therapy in the near future.[5,7,8]
EBS occurring in twins without a family history of EBS is an exceedingly uncommon occurrence, hence the report due to its rarity.
CONCLUSION
EBS is a genetically driven disorder characterised by mucocutaneous blistering, with EBS accounting for over 70% of EB cases. Diagnostic precision is achieved through electron microscopy and genetic testing. The occurrence of EBS in twins without a family history is exceedingly rare, emphasizing the importance of early genetic diagnosis. While current treatment focuses on supportive care, advancements in gene therapy hold promise for future management.
Acknowledgements
The authors acknowledge the Multidisciplinary Research Unit and Institutional Ethics Committee for Plagiarism check support.
Ethical approval:
Institutional Review Board approval is not required.
Declaration of patient consent:
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest:
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation:
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Inherited Epidermolysis Bullosa In: Sewon K, Amagai M, Bruckner A, Enk A, Margolis D, Mc Michael A, eds. Fitzpatrick Dermatology (9th ed). New York: McGraw Hill Education; 2019. p. :1011-30.
- [Google Scholar]
- Epidermolysis Bullosa-A Different Genetic Approach in Correlation with Genetic Heterogeneity. Diagnostics (Basel). 2022;12:1325.
- [CrossRef] [PubMed] [Google Scholar]
- Inherited Epidermolysis Bullosa: New Diagnostic Criteria and Classification. Clin Dermatol. 2012;30:70-7.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular Epidemiology of Hereditary Epidermolysis Bullosa in a Middle Eastern Population. J Investig Dermatol. 2006;126:777-81.
- [CrossRef] [PubMed] [Google Scholar]
- Severe Generalized Epidermolysis Bullosa Simplex in Two Hong Kong Children due to De Novo Variants in KRT14 and KRT5. Case Rep Pediatr. 2020;2020:4206348.
- [CrossRef] [PubMed] [Google Scholar]
- De Novo KLHL24 Gene Pathogenic Variants in Chinese Twin Boys with Epidermolysis Bullosa Simplex: A Case Report. Front Genet. 2021;12:729628.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates from the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016;152:1231-8.
- [CrossRef] [PubMed] [Google Scholar]
- Epidermolysis Bullosa in a Twins Infant: A Rare Case. Bali Dermatol Venereol Aesthet J. 2023;6:13-6.
- [CrossRef] [Google Scholar]




