

Translate this page into:
Red Dots and Dry Skin: What is More in the Store – A Rare Lysosomal Storage Disorder
*Corresponding author: Priya Prathap, Department of Dermatology and Venereology, Government Medical College, Thrissur, Kerala, India priya.anil.an@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Satchith N, Prathap P, Nampoothiri S, Asokan N. Red Dots and Dry Skin: What is More in the Store – A Rare Lysosomal Storage Disorder. Indian J Postgrad Dermatol. doi: 10.25259/IJPGD_34_2024
Dear Editor,
A 28-year-old male presented with complaints of dryness and scaling of legs [Figure 1a] for a 6-month duration. There was no family history of dry skin. On examination, there were ichthyotic patches with ill-defined borders on the lateral aspect of both legs. There was no sensory impairment and no nerve thickening, and the slit-skin smear was negative for acid-fast bacilli. Therefore, we made a provisional diagnosis of acquired ichthyosis. Multiple tiny discrete and confluent red to purple non-blanchable macules and papules were present on palms and lips since 6 years of age and were asymptomatic with no bleeding manifestations. They had consulted a paediatrician, and hereditary haemorrhagic telangiectasia was considered and reassured at that time. At 15 years of age, he noticed reduced sweating and experienced giddiness on exertion and intolerance to warm environments. He had pins and needle sensation of extremities. For these complaints, he was evaluated by a physician and neurologist and was treated for peripheral neuropathy with gabapentin. Since he had no relief, he was referred to a psychiatrist, who started him on clonazepam with a diagnosis of anxiety disorder. He also had a history of recurrent bilateral pedal oedema, which used to subside by limb elevation. There was no history of exertional dyspnoea, palpitation, seizure disorder and bleeding manifestations.
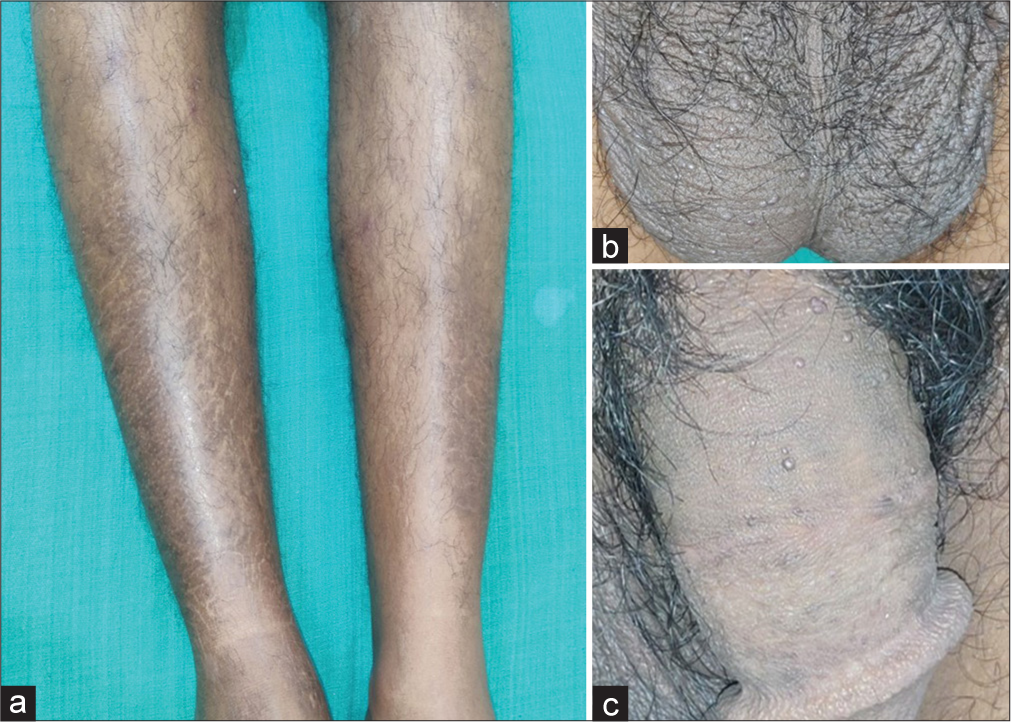
- (a) Ichthyotic macules on legs. (b) Skin-coloured papules on the scrotum. (c) Skin-coloured papules on the shaft of the penis.
Multiple tiny discrete skin-coloured papules were noted on the scrotum [Figure 1b] and shaft of the penis [Figure 1c], suggestive of angiokeratoma. There were non-blanchable multiple discrete and confluent reddish macules and papules on the trunk, arms, palms [Figure 2a] and lips [Figure 2b]. On dermoscopic examination, red lacunae intermingled with a white veil were seen on lesions on the palms [Figure 3a] and scrotum [Figure 3b], indicative of angiokeratomas.[1] This, together with his other symptoms, prompted us to review the diagnosis of angiokeratoma corporis diffusum, which is the classical cutaneous manifestation of Fabry disease. We noticed that he had coarse facial features: Prominent supraorbital ridges, bushy eyebrows, shallow midface, prominent nasal bridge, pronounced nasal angle, broad alar base and prominent earlobes. His skin was very warm to the touch due to decreased sweating. Examination with a corneometer showed decreased moisture content. There was also mild bilateral pitting pedal oedema. The rest of the systemic examination was normal.
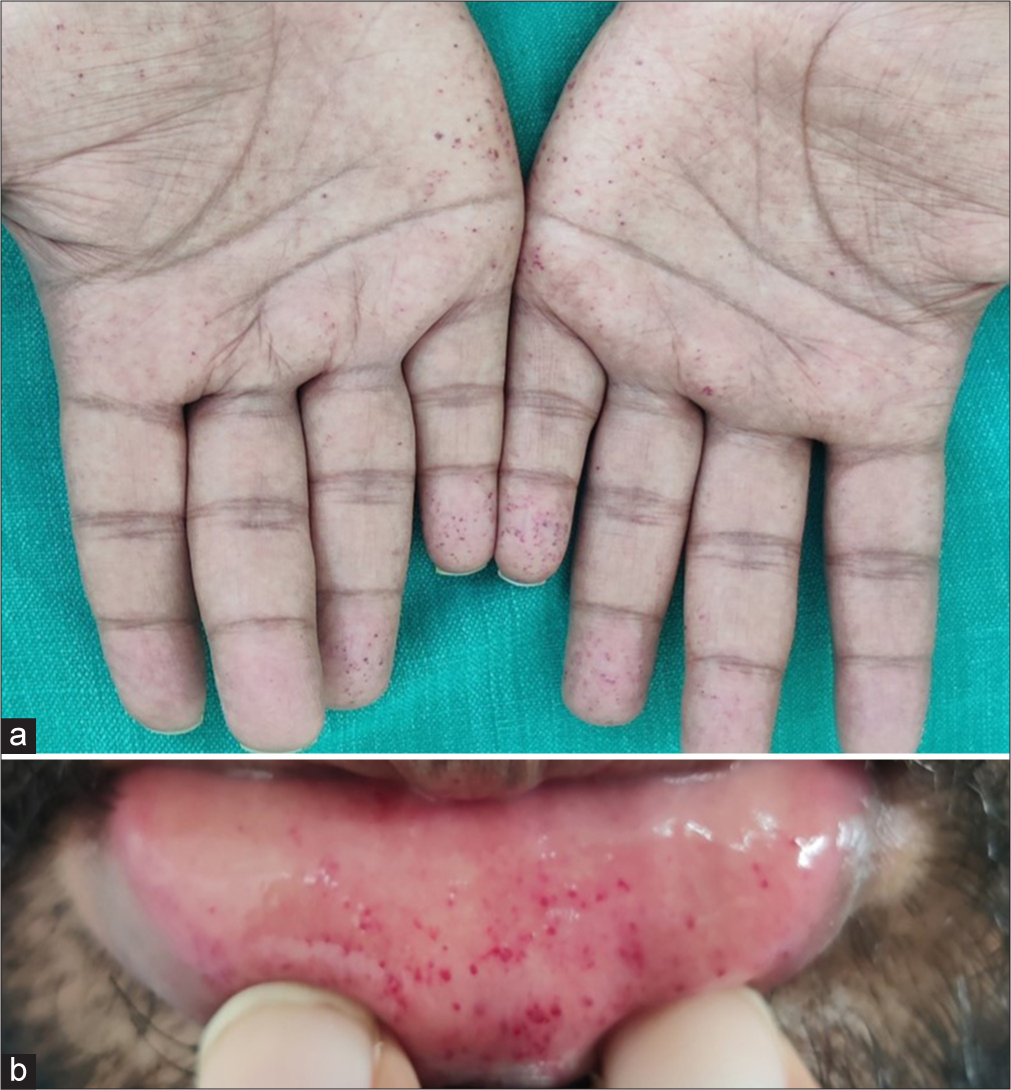
- (a) Non-blanchable reddish macules on palms. (b) Nonblanchable reddish macules on lips.
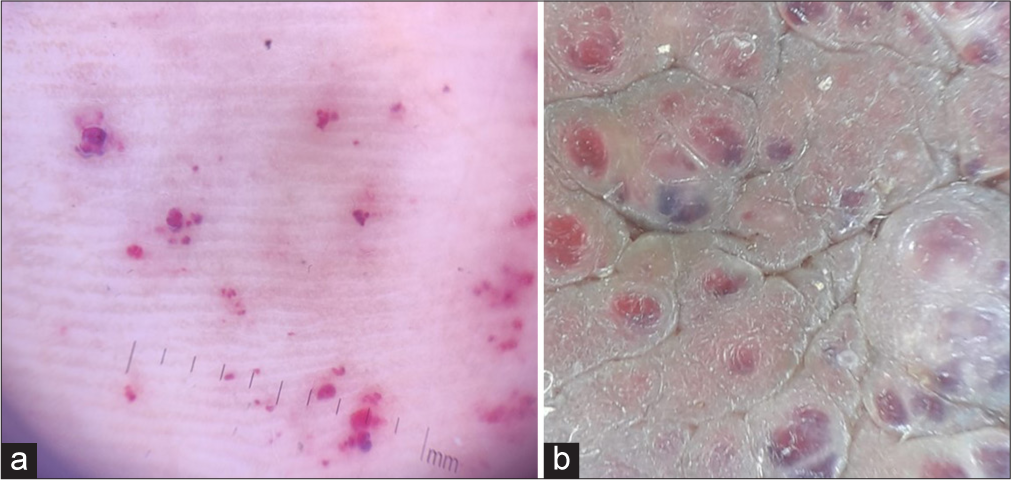
- (a) Red lacunae on dermoscopy of palmar lesions. (b) Red lacunae intermingled with a white veil on dermoscopy of scrotal lesions.
The haemogram and coagulation parameters were normal. Urine routine and 24-hour urine protein examination showed proteinuria (212.8 mg/L [<100 mg/dL]). Renal function test showed a mild increase in blood urea (33 mg/dL [20 mg/dL]) and creatinine (1.4 mg/dL [1.3 mg/dL]) as well. A 40% reduction was noted in the estimated glomerular filtration rate. The thyroid function test and ultrasound of the abdomen were normal. Doppler study of lower limbs showed subcutaneous oedema with no vascular abnormality. The postural variation was suggestive of either stage 1 lymphoedema or autonomic dysfunction. Slit-lamp examination of the eyes revealed bilateral cornea verticillata and tortuous retinal vessels were detected on fundoscopy, both of which are features of Fabry disease. The left ventricular strain pattern was identified in the electrocardiogram. Echocardiography showed concentric left ventricular hypertrophy. Magnetic resonance imaging of the brain and nerve conduction studies of all four limbs were normal. Pure-tone audiometry of both ears was normal.
We estimated the alpha-galactosidase activity, which revealed a markedly low value, 1.25 nmoL/h/mg (expected >60), which helped us to clinch the diagnosis. Genetic analysis was done, which detected a known pathogenic hemizygous variant, c.901C>T(p.Arg301Ter) in exon 6 of galactosidase alpha gene on the long arm of X chromosome, suggestive of classic Fabry disease. On the evaluation of other family members, his mother had bilateral cornea verticillata, and she, too, carried the same variant in the heterozygous state. Echocardiogram showed hypertrophic cardiomyopathy and trivial mitral regurgitation, and she had microalbuminuria.
Anderson–Fabry disease is a rare genetic X-linked lysosomal storage disorder, with deficient or absent alpha-galactosidase enzyme, which results in the accumulation of globotriacylceramide mostly in endothelial cells, kidneys, myocardium and dorsal root ganglion.[2] Incidence varies from 1/40,000 to 1/1,17,000 live births.[3] The classical cutaneous manifestation is angiokeratoma corporis diffusum. Others are cherry angioma, telangiectasia and lymphoedema. Fabry outcome survey database of 714 patients reported angiokeratoma in 66% of males and 36% of females with Fabry disease.[4]
Neuropathic pain is due to small fibre neuropathy. Hypohidrosis occurs secondary to sweat gland involvement. Cornea verticillata is due to the deposition of globotriaosylceramide in the cornea. Progressive glomerular sclerosis arises because of vascular lumen obliteration in the kidney. Diastolic dysfunction followed by concentric left ventricular hypertrophy often happens in the fourth decade. Central nervous system manifestations such as headache, vertigo, transient ischaemic attack and stroke are described.
Complications of Fabry disease include renal failure, hypertrophic cardiomyopathy, cardiac failure and stroke, which can be fatal. Mean life expectancy decreased by 20 years in males and 15 years in females.
Enzyme replacement therapy (ERT) is available in two forms: A human cell-based agalsidase alfa, given as 0.2 mg/kg biweekly as an intravenous infusion, and Chinese hamster ovary-based agalsidase beta, given as 1.0 mg/kg/intravenous biweekly.[5] Migalastat, an oral drug, is a small-molecule chaperone which facilitates enzyme trafficking to lysosomes but is effective only for amenable mutations in the alpha-galactosidase A (GLA) gene.[6] ERT was advised for our patient, but he could not afford it. He is on treatment with tablet enalapril 5 mg, a combination of taurine and acetylcysteine, which are renoprotective antioxidants, the tablet gabapentin 100 mg twice daily and emollients on legs.
Dermatologists have a crucial role in diagnosing rare metabolic diseases like Fabry, as dermatological manifestations are often the first to appear. Early initiation of ERT can improve the quality of life of the patient. Drugs for such rare diseases, if made available under an insurance scheme, would be beneficial for patients.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript, and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Dermoscopy of Solitary Angiokeratomas: A Morphological Study. Arch Dermatol. 2007;143:318-25.
- [CrossRef] [PubMed] [Google Scholar]
- Global Research on Fabry's Disease: Demands for a Rare Disease. Mol Genet Genomic Med. 2020;8:e1163.
- [CrossRef] [PubMed] [Google Scholar]
- The Effect of Enzyme Replacement Therapy on Clinical Outcomes in Male Patients with Fabry disease: A Systematic Literature Review by a European Panel of Experts. Mol Genet Metab Rep. 2019;19:100454.
- [CrossRef] [Google Scholar]
- Twenty Years of the Fabry Outcome Survey (FOS): Insights, Achievements, and Lessons Learned from a Global Patient Registry. Orphanet J Rare Dis. 2022;17:238.
- [CrossRef] [PubMed] [Google Scholar]
- Chaperone Therapy in Fabry Disease. Int J Mol Sci. 2022;8:23:1887.
- [CrossRef] [PubMed] [Google Scholar]
- Gene Therapy for Fabry Disease: Progress, Challenges, and Outlooks on Gene-Editing. Mol Genet Metab. 2021;134:117-31.
- [CrossRef] [PubMed] [Google Scholar]




