

Translate this page into:
Smooth Papules, Rough Diagnosis – A Case of Primary Systemic Amyloidosis
*Corresponding author: Yashika Jayesh Doshi, Department of Dermatology, Venereology and Leprosy, B.J. Medical College, Ahmedabad, Gujarat, India. yashikajdoshi@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Vasani PA, Doshi YJ, Shah B. Smooth Papules, Rough Diagnosis – A Case of Primary Systemic Amyloidosis. Indian J Postgrad Dermatol. 2025;3:83-5. doi: 10.25259/IJPGD_41_2024
Dear Editor,
A 55-year-old male, admitted to medical ward for generalised weakness that was present for the past 3 months, was referred to the dermatology department for asymptomatic skin lesions over the face for the past 2 months. The lesions were progressively increasing in size and number. He had no history of any systemic disease nor did he have any systemic complaints on enquiry. The patient denied bone pain, tingling sensation/numbness over hands, spontaneous bleeding from the natural orifices, bleeding gums/disproportionately more bleeding on minor trauma, recurrent infections, decreased urine output, nausea/vomiting or significant weight loss.
On cutaneous examination, he had multiple, non-tender, skin-coloured papules over nasal bridge, bilateral periorbital, retro-auricular, perinasal and perioral region [Figure 1a]. Few papules coalesced to form a firm, non-tender, skin-coloured plaque over the right cheek. Oral examination revealed macroglossia with scalloping and infiltrated papules and plaques over labial mucosa [Figure 1b and c]. Hair and nail examinations were normal. Systemic examination was unremarkable. There was no evidence of pallor, lymphadenopathy or hepatosplenomegaly.
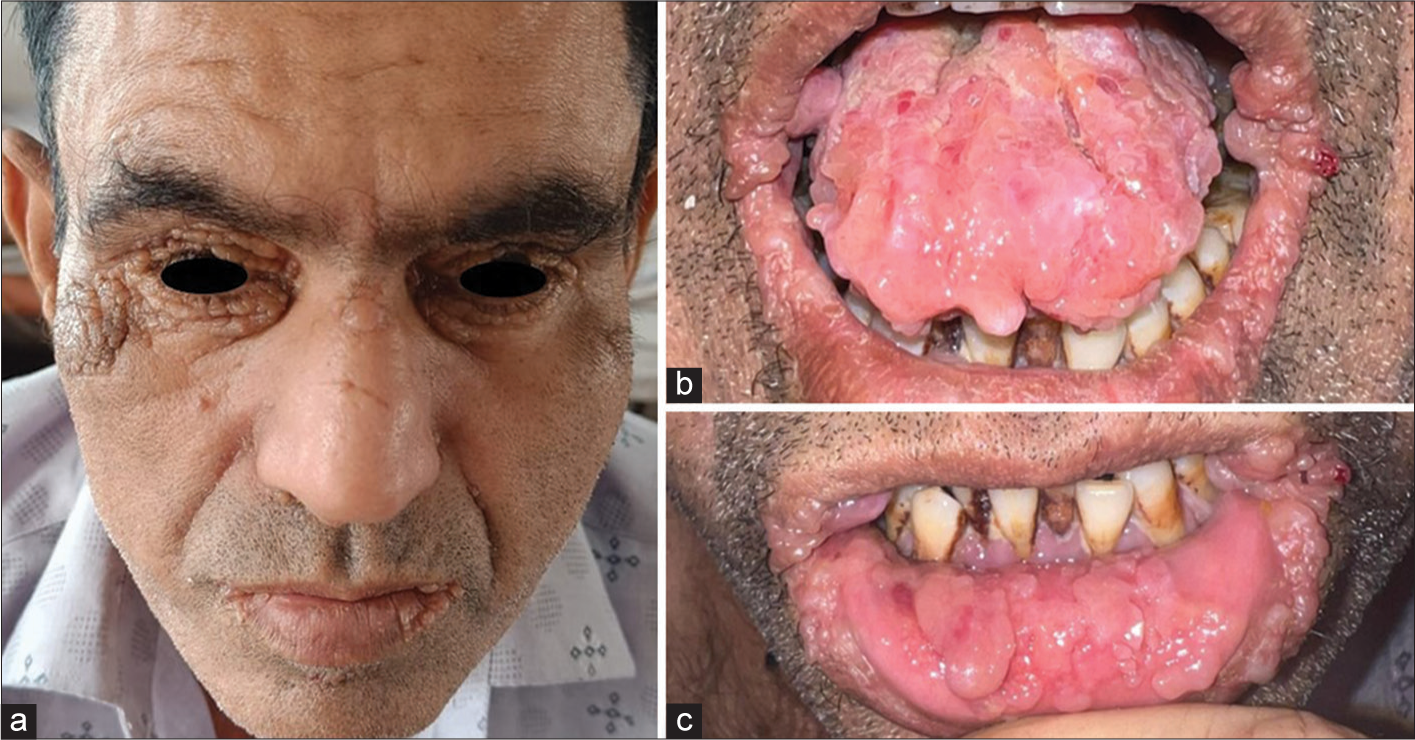
- (a) Multiple, skin-coloured papules over nasal bridge, bilateral periorbital, retroauricular, perinasal and perioral region. (b) Infiltrated papules and plaques over labial mucosa. (c) Macroglossia with scalloping.
Routine laboratory investigations included complete blood count which revealed low haemoglobin level (9.4 g/dL), rest of the parameters like erythrocyte sedimentation rate, liver and kidney function tests, urine routine and microscopic examinations were normal. The patient had raised serum calcium levels (13 mg/dL), while serum phosphate levels were normal. X-ray of long bones showed generalised osteopenia as well. Serologies for human immunodeficiency virus, hepatitis B, C and syphilis were non-reactive. His chest X-ray, electrocardiogram, 2D-echocardiography and ultrasonography of the abdomen and pelvis were normal.
A biopsy from plaque over the right cheek stained with haematoxylin-eosin showed atrophied epidermis with dense amorphous, eosinophilic, hyaline, extracellular deposits in the dermis [Figure 2a]. These deposits showed orange-red colour with Congo red stain on routine microscopy [Figure 2b] and apple green birefringence with polarised microscopy [Figure 2c]. This intrigued us to perform serum electrophoresis which revealed hypoproteinaemia and hypogammaglobulinaemia. On serum immunofixation, there was presence of a band in Lambda region, suggestive of lambda light chain disease. Bone marrow biopsy revealed diffuse and interstitial infiltration of plasma cells (>20%) and serum M protein level was 32 g/L, suggesting a diagnosis of plasma cell dyscrasia consistent with multiple myeloma. As per the CRAB criteria for multiple myeloma, which consists of (1) hypercalcaemia: serum calcium >11 mg/dL, (2) renal insufficiency: serum creatinine >2 mg/dL or creatinine clearance <40 mL/min, (3) anaemia: haemoglobin <10 g/dL and (4) lytic bone lesions or osteopenia. Since this patient was fulfilling 3 out of 4 parameters in CRAB criteria, a diagnosis of primary systemic amyloid light-chain (AL) amyloidosis secondary to multiple myeloma was made. Serum lactate dehydrogenase value was 425 U/L and serum b2 microglobulin level was 4.2 mg/L. Urine failed to show Bence–Jones protein on electrophoresis. The patient was referred to the haematooncologist. He was advised cyclophosphamide, bortezomib and dexamethasone chemotherapy but did not agree to comply with the provided treatment.
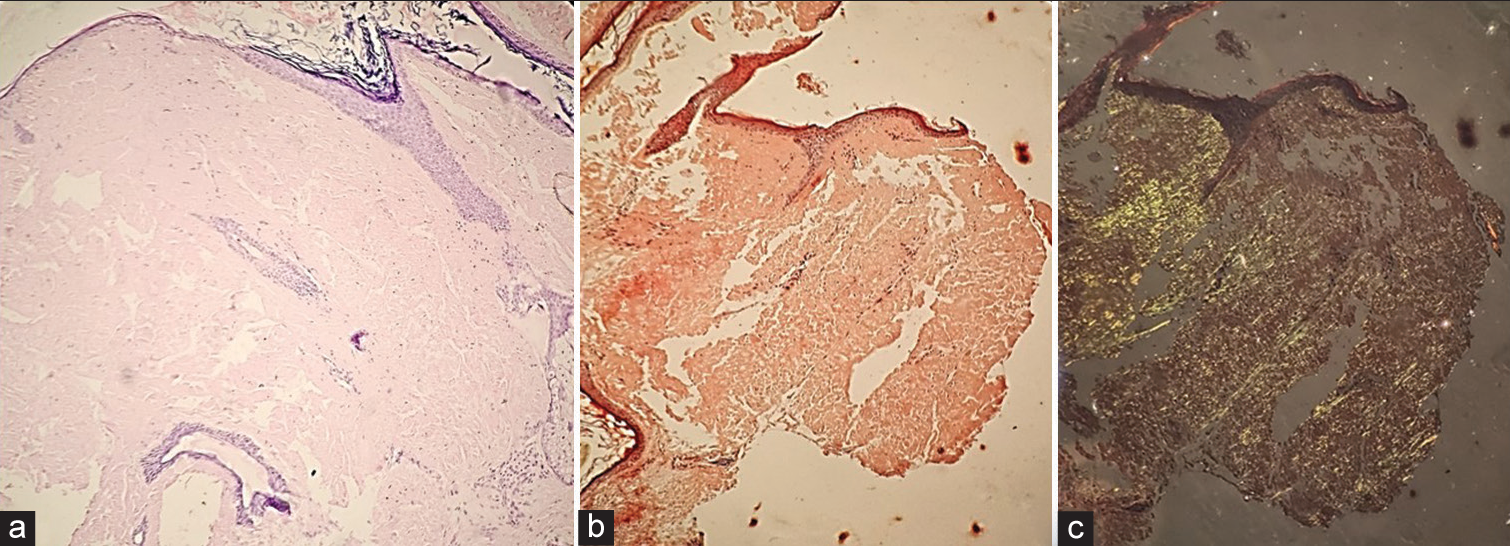
- (a) Histopathology showing atrophied epidermis with dense, amorphous, eosinophilic, hyaline extracellular deposits in the dermis (H&E ×20). (b) Amyloid deposits showing orangered colour with Congo red stain on routine microscopy (H&E ×20). (c) Amyloid deposits showing apple-green birefringence with Congo red on polarised microscopy (H&E ×20). H&E: Haematoxylin & Eosin.
Amyloidosis is a rare group of disorder, characterised by extracellular deposition of amyloid protein. Primary systemic amyloidosis may present with protean manifestations due to multiple organ involvements. While renal involvement, cardiomyopathy, carpal tunnel syndrome and peripheral neuropathy are common, it is relatively unusual for a patient with systemic amyloidosis to present with just fatigue and skin involvement.[1]
Primary systemic amyloidosis can present with mucocutaneous manifestations in 30–40% of cases, where the classical cutaneous features include periorbital purpura (raccoon eyes), infiltrated papules and macroglossia.[2] Atypical cutaneous manifestations such as haemorrhagic bullae, dissecting haematomas, scleroderma-like changes, cutis laxa-like changes, blue skin tint, follicular hyperkeratotic spicules, diffuse alopecia, chronic paronychia and cutis verticis gyrata have been reported in the past.[1,3] Cutaneous features depend upon the site of amyloid deposition. While petechiae, pinch purpura and ecchymosis are said to be common presentations due to infiltration of blood vessel walls by amyloid deposits, they were not observed in our case.[4] Systemic involvement is frequent in cases with AL amyloidosis, which includes renal involvement in 70%, cardiomyopathy in 60%, peripheral neuropathy in 20% and gastrointestinal in 70% of cases.[5] However, there was no systemic involvement in the present case. The various differentials that can be considered and their presenting features have been described in Table 1.
| Disease | Age of onset | Presentation | Histopathology |
|---|---|---|---|
| Papular mucinosis | Begins in infancy (cutaneous mucinosis of infancy) or adulthood (discrete papular form, acral persistent papular form, pure nodular form) | • 2–5 mm papules, few to hundreds, distributed in symmetrical pattern • Face is spared • Can be associated with HIV or paraproteinaemia |
Mucin deposition in the dermis (Alcian blue positive, congo red negative) |
| Nodular amyloidosis | Rare disease, most reported cases have adult onset | • Single or multiple waxy nodules or infiltrated plaques, most commonly on the trunk or extremities • Immunoglobulin γ light chains and β2 microglobulin produced by plasma cells within the vicinity of deposits • No evidence of plasma cell proliferation in the bone marrow • Can be associated with Sjogren syndrome |
Amorphous, eosinophilic, hyaline, extracellular amyloid deposits in the dermis. (Congo red positive) |
| Lipoid proteinosis | Early age of onset | • Usually presents with hoarse voice, ice pick like scars, Monilial blepharosis, neurological manifestations • Positive family history |
Hyaline deposits in the dermis (Alcian blue positive, PAS positive, congo red negative) |
| Sebaceous hyperplasia | Usually presents in adulthood/elderly | • Single or multiple, yellowish, occasionally telangiectatic papules with central dell usually on the central and upper face | Enlarged sebaceous lobules |
| Eruptive xanthoma | Adolescent/adult onset | • Erythematous to yellow papules, 1–5 mm in diameter, usually on extensor surfaces of the extremities, hands and buttocks. | Lipid deposits in reticular dermis, foam cells seen. |
HIV: Human immunodeficiency virus, PAS: Periodic acid–Schiff
Apart from fatigue, the presence of classical cutaneous lesions was the only clue leading to the diagnosis of multiple myeloma in our case. This reaffirms the role of a dermatologist in the multimodality management of primary systemic amyloidosis. Diagnosing these patients in time provides an opportunity for early initiation of treatment before further systemic involvement, thus prolonging survival in such diseases that usually have a guarded prognosis.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Case Report: One Case of Primary AL Amyloidosis Repeatedly Misdiagnosed as Scleroderma. Medicine (Baltimore). 2017;96:e8771.
- [CrossRef] [Google Scholar]
- Cutaneous Amyloidoses and Systemic Amyloidoses with Cutaneous Involvement. Eur J Dermatol. 2010;20:152-60.
- [CrossRef] [Google Scholar]
- Unusual Milia Amyloidosis as Initial Signs of Multiple Myeloma-associated Systemic Amyloidosis. Int J Dermatol. 2013;52:981-2.
- [CrossRef] [Google Scholar]




